

烷烴與多種自由基(如Cl、O(3P)、OH、NH和CN)在低溫區發生反應時,速率常數與溫度呈現出特殊的負相關性,即隨著溫度的升高反應速率常數反而降低。
為了解釋這一反常現象,美國能源部Sandia國家實驗室的Georgievskii等人借助CASPT2、QCISD(T)等高精度量化方法對氰自由基(CN)與乙烷分子的反應機理進行研究,并分析了這類反應的動力學性質。研究證實,在低溫區(<200k)這類自由基反應體系的決速步驟是通過弱相互作用(范德華力)形成復合物,而在高溫區的決速步驟則涉及到化學鍵的斷裂和生成。通過仔細考察反應過程中涉及的兩種過渡態機制,Georgievskii等人提出一種雙過渡態模型,其理論計算的結果準確地重現了實驗中觀察到的特殊溫度效應曲線。
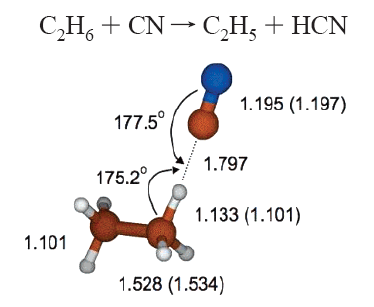
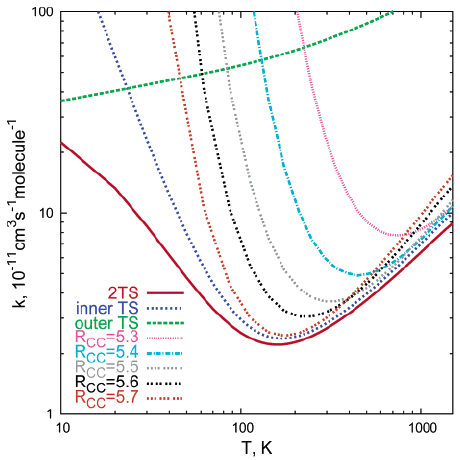
圖1、CASPT2方法優化得到的氰自由基與
乙烷分子反應的過渡態結構圖2、雙過渡態模型與其它過渡態模型的結果對比
Ref: Y. Georgievskii; S. J. Klippenstein, J. Phys. Chem. A, 2007, 111, 3802-3811.
使用軟件:Molpro
近年來疊氮自由基(N3)與氟原子(F)的反應機理得到了廣泛的關注,該反應體系在新型激光器的研制方面有著潛在的用途。
中科院化學所邊文生等人用CASSCF和MRCI方法對N3+F反應體系進行研究,通過掃描單重態和三重態的勢能曲線確定了兩態之間的最低能量交叉點,從而澄清了疊氮自由基與氟原子的反應機制。計算結果表明,該反應體系的單重態和三重態之間存在著絕熱和非絕熱兩種轉化方式,其中絕熱反應方式(圖1, 式i)將占主導優勢,這與實驗觀測到的現象相吻合。
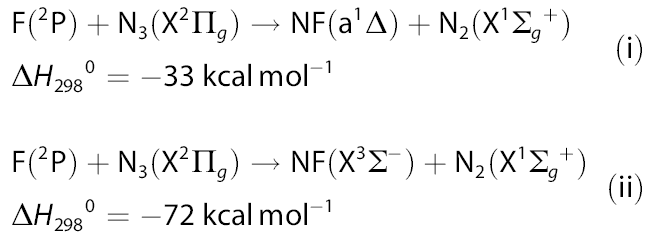
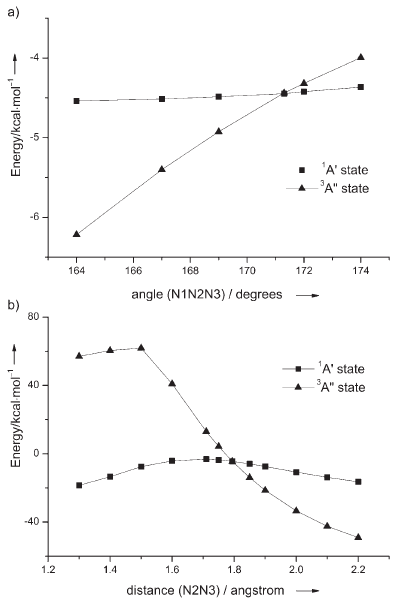
圖1、氟原子與疊氮自由基反應的兩種產物
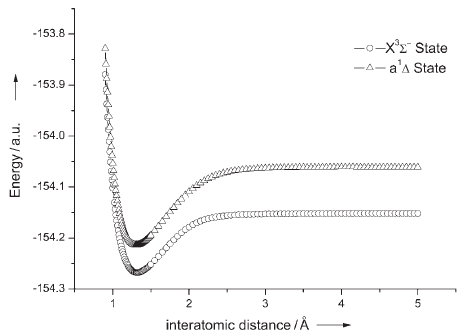
圖2、產物NF分子的單重態和三重態勢能曲線
圖3、疊氮自由基N3的單重態和三重態勢能曲線
Ref: H. T. Ma; X. J. Liu; W. S. Bian; L. P. Meng; S. J. Zheng, Chemphyschem, 2006, 7, 1786-1794.
使用軟件:Molpro
QM/MM方法混合使用量子力學和分子力學,能夠顯著降低計算量,近年來已成為模擬酶催化反應的一種重要手段。由于酶催化反應涉及的體系通常比較大,即使采用了QM/MM方法,需要用量子力學處理的原子數仍然比較多,因而目前多采用精度相對較低的計算方法,如半經驗方法或密度泛函理論(DFT)。在處理較大分子體系的時候半經驗方法是一個不錯的選擇,但是其精度相對較低,計算誤差有可能達到十幾個kcal/mol-1甚至更高。相對而言,DFT方法能夠在一定程度上提高準確性,但是對于色散力等關鍵的弱相互作用仍然難以處理,因此DFT方法通常會低估反應能壘約幾個千卡。
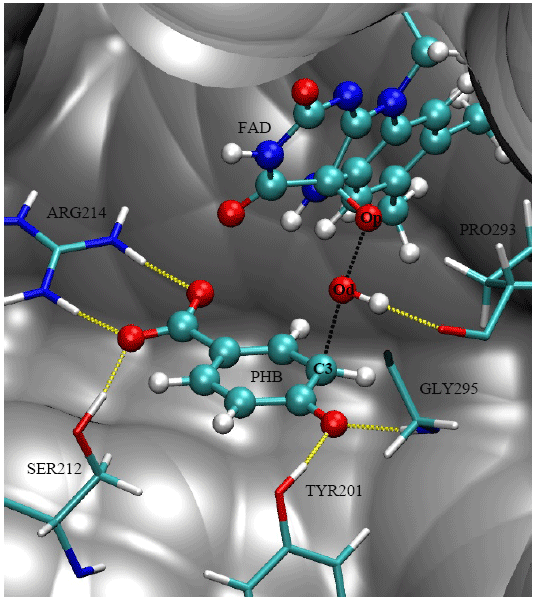
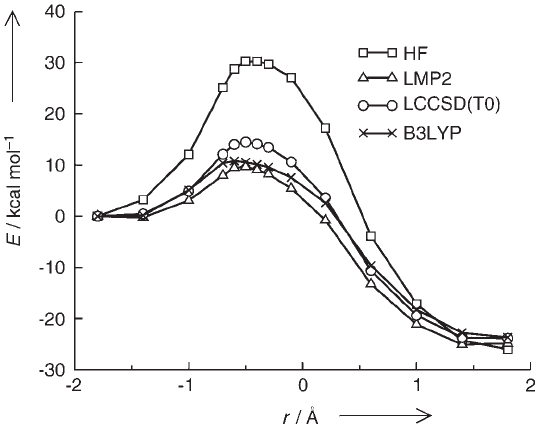
圖1、對羥苯甲酸羥化酶PHBH的過渡態結構 圖2、不同計算方法掃描得到的CM體系的勢能曲線對比
最近,英國布里斯托大學的Mulholland、德國馬普協會煤化所的Thiel等人采用QM/MM方式對分支酸變位酶(chorismate mutase,CM)和對羥苯甲酸羥化酶(para-hydroxybenzoate hydroxylase,PHBH)在溶液環境中的反應過程進行模擬,計算過程中這兩種酶催化體系分別含有7千和2萬3千個原子。他們用Molpro軟件的B3LYP、LMP2和LCCSD(T0)等高精度計算方法處理QM部分的原子,成功地預測了這兩種酶在催化反應過程中的活化焓和自由能。計算結果表明,LCCSD(T0)的計算結果最為準確,其預測值與實驗觀測值幾乎完全吻合,而B3LYP和LMP2方法在不同程度上都低估了酶催化反應的活化能壘。
Ref: F. Claeyssens; J. N. Harvey; F. R. Manby; R. A. Mata; A. J. Mulholland; K. E. Ranaghan; M. Schutz; S. Thiel; W. Thiel; H. J. Werner, Angew. Chem. Int. Ed., 2006, 45, 6856-6859.
使用軟件:Molpro
樹枝狀聚苯乙炔(Phenylacetylene Dendrimer,PAD)是一種帶有多個發色團的支化大分子,在光照激發之后各個聚合單元之間能夠迅速而有效地傳遞能量。傳統的理論認為間位連接的苯乙炔單元會阻塞電子態的離域,因此PAD的能量傳遞將被局限在樹枝狀分子的分枝上。
美國伊利諾斯大學的Martinez和Bardeen等人認為這種理論僅僅考慮了基態分子的電子結構,而對于激發態的PAD來說,情況可能會有很大差異。為此他們采用CASSCF和CASPT2等方法對PAD分子進行研究,結果表明,盡管PAD各單元之間在處于基態時耦合作用非常微弱,但是在處于激發態時由于構型的變化耦合作用將會大幅度增強。在此基礎上,Martinez和Bardeen等人對傳統的Forster能量傳遞模型提出了修正建議。
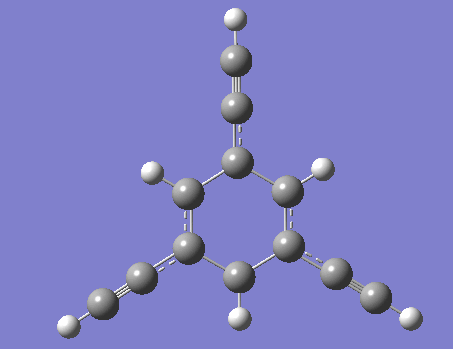
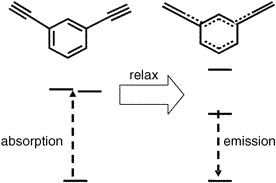
圖1、三乙炔基苯處于激發態時的最優構型 圖2、PAD單元的激發、馳豫和發射示意圖
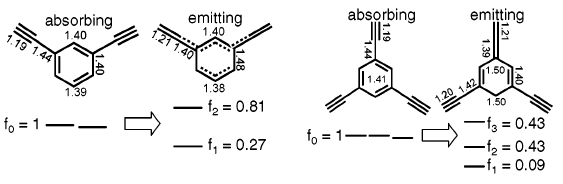 圖3、苯乙炔單元在吸收、發射狀態的鍵長和躍遷偶極矩
圖3、苯乙炔單元在吸收、發射狀態的鍵長和躍遷偶極矩
Ref: K. M. Gaab; A. L. Thompson; J. J. Xu; T. J. Martinez; C. J. Bardeen, J. Am. Chem. Soc., 2003, 125, 9288-9289.
使用軟件:Molpro
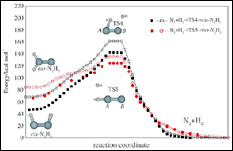
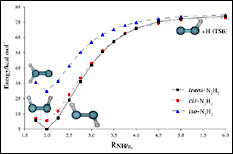
眾所周知,由于氮氣的氫化反應具有非常高的活化能壘,在沒有催化劑存在時,這個反應只有在極端條件下才可能發生。目前人們尚未完全掌握氮氣加氫的詳細反應機制,僅知道氮氣分子與第一個氫分子的反應是整個氫化過程中的決速步驟,因此,葡萄牙科英布拉大學Varandas等人利用MCSCF/aug-cc-pVQZ方法對偶氮烯(N2H2)分子進行高精度計算,詳細地分析了N2H2分子的cis、trans和異構模型的最優構象,并計算了這幾種構型之間相互轉變的過渡態能壘,以求獲得精確而全面的基態勢能曲線,從而有助于了解氮氣氫化過程的反應機理。
 |
 |
| 圖1.氮氣氫化過程的兩種反應方式 | 圖2.偶氮烯三種構型的分解反應路徑 |
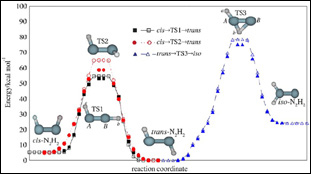 |
| 圖3.偶氮烯的cis、trans和iso三種構型的最優構象,以及三者之間相互轉變的過渡態結構 |
Ref: M. Biczysko; L. A. Poveda; AJC Varandas, Chem. Phys. Lett., 2006, 424, 46-53.
使用軟件:Molpro