

QM/MM方法混合使用量子力學和分子力學,能夠顯著降低計算量,近年來已成為模擬酶催化反應的一種重要手段。由于酶催化反應涉及的體系通常比較大,即使采用了QM/MM方法,需要用量子力學處理的原子數仍然比較多,因而目前多采用精度相對較低的計算方法,如半經驗方法或密度泛函理論(DFT)。在處理較大分子體系的時候半經驗方法是一個不錯的選擇,但是其精度相對較低,計算誤差有可能達到十幾個kcal/mol-1甚至更高。相對而言,DFT方法能夠在一定程度上提高準確性,但是對于色散力等關鍵的弱相互作用仍然難以處理,因此DFT方法通常會低估反應能壘約幾個千卡。
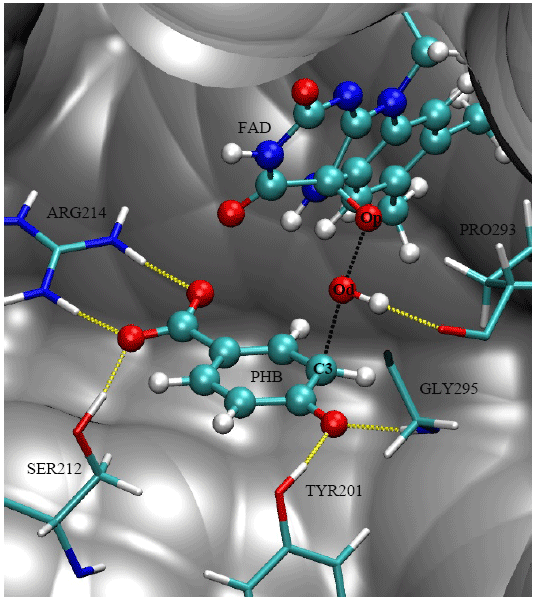
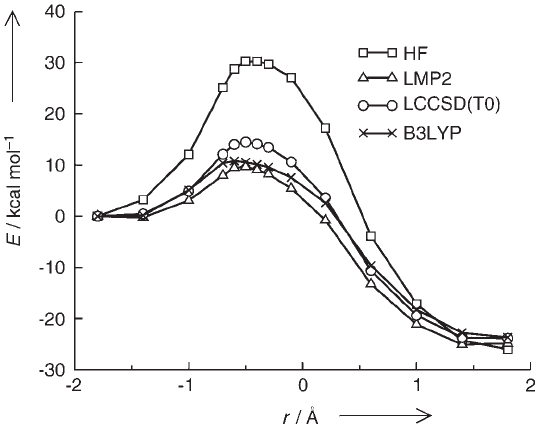
圖1、對羥苯甲酸羥化酶PHBH的過渡態結構 圖2、不同計算方法掃描得到的CM體系的勢能曲線對比
最近,英國布里斯托大學的Mulholland、德國馬普協會煤化所的Thiel等人采用QM/MM方式對分支酸變位酶(chorismate mutase,CM)和對羥苯甲酸羥化酶(para-hydroxybenzoate hydroxylase,PHBH)在溶液環境中的反應過程進行模擬,計算過程中這兩種酶催化體系分別含有7千和2萬3千個原子。他們用Molpro軟件的B3LYP、LMP2和LCCSD(T0)等高精度計算方法處理QM部分的原子,成功地預測了這兩種酶在催化反應過程中的活化焓和自由能。計算結果表明,LCCSD(T0)的計算結果最為準確,其預測值與實驗觀測值幾乎完全吻合,而B3LYP和LMP2方法在不同程度上都低估了酶催化反應的活化能壘。
Ref: F. Claeyssens; J. N. Harvey; F. R. Manby; R. A. Mata; A. J. Mulholland; K. E. Ranaghan; M. Schutz; S. Thiel; W. Thiel; H. J. Werner, Angew. Chem. Int. Ed., 2006, 45, 6856-6859.
使用軟件:Molpro