
Q-CHEM
![]()
Q-Chem是一款功能齊全的從頭算量子化學程序包,最早由諾貝爾化學獎獲得者John A. Pople主導開發。全面支持從DFT/HF到各種高級的post-HF相關能計算方法。并提供廣泛的解決方案,適用于不同的研究目標,如單分子磁體的自旋軌道耦合效應、高通量計算、酶的量子力學/分子力學(QM/MM)研究等。被廣泛用于處理工業界、學術界和國家實驗室的各類理論模擬研究,在理論化學、藥物設計、材料科學、生物化學以及相關領域的教學和研究中發揮了極大作用。可以包括研究分子結構、化學反應、分子振動、電子光譜、NMR譜和溶劑化效應等。
Q-Chem自1999年第一個版本1.2發布至今,已經陪伴廣大科研用戶26年了。自1999年以來,Q-Chem引領量子化學計算的潮流,不斷突破科技邊界,26年的專注研發,每年至少三次的版本迭代,Q-Chem始終站在量子化學軟件的最前沿。 從1.2版本到最新的6.2,Q-Chem的每一次版本升級都是對科學的一次深刻致敬。
Q-Chem目前最新版本是6.2,是該軟件包的第七個主要版本,包含多項改進和新特性。一些新增特性包括:
*自然增強軌道(Natural Auger Orbitals)用于CVS-EOM方法的奧杰衰減、ICD和相關過程。
*ACP-EOMIP-CCSD用于部分奧杰衰減寬度的計算。
* EOM-CCSDT用于電子能量和自旋-翻轉態的研究。
* TDKS的偶極濾波。
* DFT/CIS半經驗方法,包括用于X射線光譜學的新的參數化。
* 1C-NOCIS的推廣到雙電子開放殼層單重態。
* 使用IAOs的原子多極矩計算。
* RT-NEO及其變體,NEO多態DFT(NEO-MSDFT),SCS-RIMP2和SOS-OOMP2用于NEO方法。
密度泛函理論(DFT):支持LDA、GGA、meta-GGA泛函,以及這些泛函的混合、范圍分離混合和雙重混合版本。可以評估基態和激發態的單點能量、幾何優化、振動頻率計算等。
電子相關性:提供處理電子相關效應的先進工具,如M?ller-Plesset微擾理論和耦合簇理論。對于強相關系統,提供特殊處理方法,包括CASSCF、耦合簇價鍵理論、選擇性CI、RAS-CI、自旋翻轉和變分2-RDM方法。
激發態方法:提供多種方法研究電子激發態,包括CIS、TD-DFT、NOCI、EOM-CC和ADC。這些方法可以模擬光譜特性、電荷和能量轉移以及非絕熱動力學。
溶劑化和嵌入:提供多種模型化溶劑化系統的解決方案,包括隱式溶劑模型和有效片段勢方法。還包括QM/MM和密度嵌入等多種嵌入方法。
光譜學建模:提供多種工具模擬不同類型的光譜,包括紅外和拉曼光譜、紫外-可見光譜、X射線光譜、光電子能譜、核磁共振和非線性光譜。
分子間相互作用:提供基于絕對局域分子軌道的能量分解分析,以及對稱性適應的微擾理論(SAPT)和擴展的多體版本
(XSAPT)來計算和分析分子間相互作用。
化學反應:提供幾何優化、勢能面掃描、過渡態搜索和內在反應坐標跟蹤的方法,適用于化學活性、熱化學和化學動力學的研究。
分子動力學:Q-Chem可以執行從頭算分子動力學(AIMD),包括NVE和NVT熱采樣,以及準經典分子動力學(QMD)。還包括Tully的最少交換面跳躍(FSSH)方法,有效處理非絕熱系統。
分子動力學:Q-Chem可以執行從頭算分子動力學(AIMD),包括NVE和NVT熱采樣,以及準經典分子動力學(QMD)。還包括Tully的最少交換面跳躍(FSSH)方法,有效處理非絕熱系統。
& Q-Chem 4.1新功能
- - 高度并行化(OpenMP、MPI)和有效多核兼容,實現HF、DFT和CCSD計算的高效運行;
- - 使用最新技巧描述反應分子橋聯的非平衡條件的T-Chem量子輸運代碼;
- - M06, M08 and M11系列泛函的TDDFT計算;
- - 擴展的凍結字符串方法,尤其是可用于過渡態結構細化的近似Hessian的構建;
- - 擴展絕對定域分子軌道基能量分解分析計算到非限制性體系;
- - XYGJ-OS分析能量梯度;
- - 基于SCF計算的顯式極化(XPol)單體方法,計算多體極化效應達線性標度;
- - 用于分子間相互作用能量分解分析的基于“SAPT0”水平的對稱性匹配微擾理論的計算;
- - XPol與SAPT結合的XSAPT方法計算大分子簇內串分子間相互作用;
- - 研究激發態的RAS-n-SF方法
- - 勢能面掃描;
- - 具高密度和低密度極限間連續過渡的最新精確的LSD 相關泛函;
- - 不破壞Kohn-Sham對稱性的計算定域原子磁矩的新方法;
- - 計算包括靜態相關效應的Mayer型鍵序。
& Q-Chem4.0在取得巨大成功的3.2版的優異功能的基礎上,增加了:
- - 用于激發態結構優化的TDDFT的解析梯度和解析二階導數計算
- - 復雜位能面的IRC搜尋,
- - 范圍分割和擴散校正的DFT泛函,
- - 更快的DFT、HF和耦合簇(CC)計算方法,
- - 更多選擇的激發態、溶劑化效應和電荷遷移的計算方法,
- - 更有效的處理大分子體系的QM/MM方案,
- - 大分子體系的激發態計算的最大重疊方法,
- - 雙電子性質的分析,
- - 多核系統和GPU使用的內存共享。
& Q-Chem 的特色
- 1. 超快速DFT 計算(為一般DFT 計算速度的3 倍)
- 2. 超快速MP2 計算(為一般MP2 計算速度的3-10 倍,與HF 的計算速度相當)
- 3. 第一個線性標度的NMR 化學位移計算
- 4. 適于自由基和激發態計算的新的DFT 和post-HF 計算方法
- 5. 輸入簡便的QM/MM 雜化方法計算
- 6. 新一代連續溶劑化計算模型(sm8, cosmo)
- 7. 多核系統和GPU 使用的內存共享。
& 基態自洽場方法
1. Hartree-Fock 方法
- - 限制性,非限制性,和限制性開殼層形式
- - 用于結構優化的解析一階導數
- - 用于諧振頻率分析的解析二階導數
2. 密度泛函理論
- - 局域泛函和梯度校正泛函
- - 交換泛函:Slater,Becke’88,Perdew’91,Gill’96,Gilbert-Gill’99,Handy -Cohen OPTX
- - 關聯泛函:VWN5,Lee-Yang-Parr,Perdew-Zunger’81,Perdew’86,Wigner, Perdew’91
- - EDF1 交換-關聯泛函。
- - 用戶定義的交換-關聯泛函。
- - HF-DFT 混合泛函:B3LYP,B3PW91,B3LYP5
- - 用戶定義的混合泛函。
- - 基于數值格點的數值積分方案:SG-0 標準網格,SG-1 標準網格,Lebedev 和Gauss-Legendre 角向積分方案
- - 用于結構優化的解析一階導數
- - 用于諧振頻率分析的解析二階導數
3. 線性標度方法
- - 傅立葉變換庫侖方法
- - 連續快速多極方法
- - 線性標度HF 交換方法
- - 基于格點的線性標度積分,用于交換-關聯泛函求值
- - 線性標度NMR 化學位移
4. AOINTS 包用于雙電子積分
- - 結合了高性能積分技術的最新進展;COLD PRISM;J-矩陣引擎。
5. SCF 改進
- - in-core 和直接SCF 的最優混合
- - DIIS
- - 初始猜測方案:重疊球平均原子密度,廣義Wolfsberg-Helmholtz,從小基組投影,芯哈密頓量的猜測
- - SCF 波函的穩定性分析
& 最大重疊方法
- - Fock 矩陣的直接最小化
- - 極化原子軌道對分子優化的最小基
& 基組
- 1. 高斯基組
- 2. 贗勢基組
- 3. 用戶定義的基組和贗勢
- 4. 基組重疊誤差(BSSE)校正
& QM/MM
- 1. 到CHARMM 的接口
- 2. ONIUM
- 3. 更有效地處理大分子體系的QM/MM 方案
& 基于波函的電子關聯處理
1. MP 微擾理論
- - 限制性,非限制性,和限制性開殼層形式
- - 直接和半直接方法計算能量
- - 半直接方法的解析梯度,用于限制性和非限制性形式
- - 在MP3,MP4 和MP4SDQ 方法的解析梯度計算中處理凍芯軌道
2. 局域MP2 方法
- - 根據物理圖象截斷完全MP2 的能量表達式,從而減少計算量
- - 減少計算量相對于分子尺寸的標度,近似為兩倍,卻不明顯丟失精度。
- - 應用外推PAO 用于局域校正
- - 可以使用分子中的雙原子和分子中的三原子技術
3. RI-MP2
- - 比MP2 和局域MP2 快十倍
4. 耦合簇方法
- - CCSD:能量,以及作為能量有限差分的梯度
- - EOM-XX-CCSD;XX = EE, EA, IP, SF,能夠靈活處理自由基,鍵的斷裂,以及對稱破缺問題
- - 耦合簇能量的非迭代校正:三級校正CCSD(T),三級和四級校正CCSD(2)
- - 廣泛應用分子點群對稱性,以改善效率
- - 二次雙激發耦合簇
- - QCISD,QCISD(T)和QCISD(2)用于能量
- - DIIS 用于收斂加速
- - 凍芯近似,用于增加可處理體系的尺寸
5. 優化軌道的耦合簇方法
- - 優化軌道的雙激發耦合簇(OD):可避免人為的對稱破缺問題;優化平均場參考軌道使能量最小;Brueckner
- - 耦合簇;OD,OD(T),和OD(2)的能量及梯度
- - 優化價軌道的耦合簇方法(VOD):傳統CASSCF 方法的耦合簇近似;在價活性空間利用截斷的OD 波函;
- - 比CASSCF 有更少的磁盤空間需求和更小的體系標度,可處理較大體系;VOD,VOD(T),VQCCD,和VOD(2)的能量及梯度
& 激發態方法
1. 支持的計算類型
- - 垂直激發吸收譜
- - 通過激發態能量的有限差分,進行激發態的結構優化
- - UCIS 和RCIS 進行激發態的振動分析
- - 自旋反轉DFT
2. CIS 方法
- - 從Hartree-Fock 基態波函計算激發態:獲得定性的單電子激發態;結構與頻率與基態Hartree-Fock 結果有可比性
- - 高效的直接算法用于計算閉殼層和開殼層體系的能量、解析梯度和二階導數
- - XCIS 用于二重和四重態計算
- - 雙激發微擾校正CIS(D),可使CIS 誤差減少兩倍或更多,接近于MP2
3. TDDFT
- - 從Kohn-Sham 基態波函計算激發態能量
- - 對于低位價激發態,TDDFT 比CIS 有相當大的改善,但只有相近的計算量
- - 提供激發態中關聯效應的內在圖像
- - 自由基的低位價激發態,比CIS 有相當大的改善
- - 自旋反轉密度泛函理論(SFDFT):把TDDFT 推廣到低位價激發態之外;可用于鍵斷裂的過程,以及自由基和雙自由基體系。
4. 基于耦合簇的激發態方法
- - EOM-CCSD
自旋反轉激發態方法:改善了雙、三自由基體系的處理;結合單行列式波函處理鍵斷裂問題;可用于OD和CCSD 理論級別
- - OOD 方法:與CCSD 激發態方法有幾乎相同的數值性能;比TDDFT 精度更高,到計算量更昂貴
- - EOM-VOOD 方法:類似于EOM-CCSD,但使用VOOD 方案
- - 激發態特性計算:躍遷偶極矩和結構
5. 分解分析
- - 顯示電子躍遷的工具,用于把電子躍遷分類為價躍遷、Rydberg 躍遷,混合躍遷,或電荷轉換
& 特性分析
1. 自動結構優化和過渡態優化
- - 具有一般約束的結構優化:可施加于鍵角,二面(扭轉)角或平面外的彎曲;直角坐標中凍結原子;約束不一定要加在初始結構上
- - 用約化內坐標保證迅速收斂,避免初始力常數矩陣
- - 優化使用笛卡爾,Z-矩陣或離域內坐標
- - 本征矢跟蹤算法,用于過渡態和最小化
- - GDIIS 算法用于最小化:使到平衡結構的收斂獲得極大加速
- - 內反應坐標跟蹤:沿著反應路徑的連續平衡結構和過渡態
2. 振動光譜
- - 自動調用解析和數值二階導數
- - 紅外和拉曼強度
- - 輸出標準的統計熱力學信息
- - 同位素替換,用于與實驗進行比較
- - 非諧性校正
3. NMR 屏蔽張量
4. 自然鍵軌道NBO 分析
5. Stewart 原子
- - 從分子密度重新獲得原子特性
- - Q-Chem 用單位分解方法計算這些值
6. 動量密度
7. Intracules
- - 獨特的雙電子函數,提供分子中庫侖能和交換能關于位置和動量的最詳盡信息
8. 分子中的原子
- - 利用免費的AIMPAC 進行AIM 分析
9. 溶劑模型
- - SM8
- - COSMO
- - 簡單的Onsager 反應場模型
- - Langevin 偶極模型
- - SS(V)PE:一種新的電解質連續模型
10.基于Dirac-Fock 理論的相對論能量校正
11.對角絕熱校正
- - 計算Born-Oppenheimer 對角修正,研究核與電子運動絕熱距離的分解
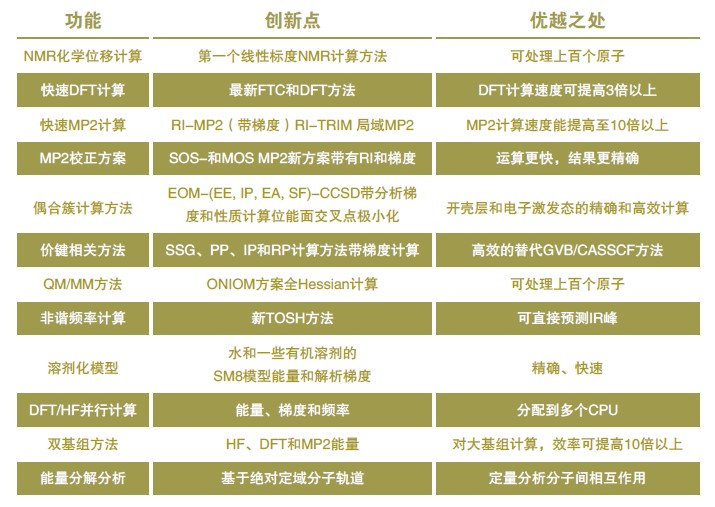
& Q-Chem 創新點
& Q-Chem效用示例
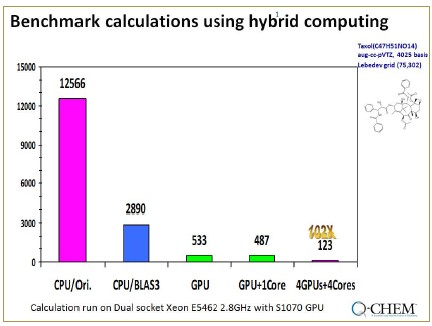
- Linux機群的快速DFT計算
對于有103個原子的紫杉醇分子,在有8CPU的Xeon機群上進行BLYP/6-31(df,pd)等級(1925個基函數)的能量和梯度計算,使用Q-Chem僅需28分鐘。
- MP2能達到HF的計算速度
高效的RI算法使中等的大基組的MP2計算更為有效。左圖是丙氨酸四肽相對構象能(有27個穩定結構)的比較的示例。
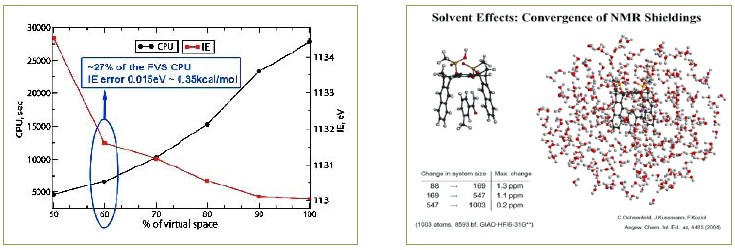
- 線性標度NMR計算
右圖和表格給出計算的分子的NMR位移相對于環繞的水分子的數目的收斂性。這是迄今進行的最大體系的NMR計算,在HF/6-31G**等級共有3000個基函數。
- 新發現
Q-Chem獨有的EOM-(EE, IP, EA, SF)-CCSD方法能有效地描述雙或三自由基的多個組態。使用該方法發現了首個具有開殼層雙重態基態的有機分子的實例,DMX三自由基的三個不成對電子呈反鐵磁性式偶合。